
International Journal of Scientific & Engineering Research, Volume 6, Issue 3, March-2015 477
ISSN 2229-5518
In silico prediction and threading based epitope mapping of OmpA-like outer membrane Leptospiral Lipoprotein Loa22
Angela Asir Ramasamy Victor1$, Sunil Abraham1$, Jebasingh Tennyson1, Nityananda Pradhan2*
Abstract — Loa22 is OmpA-like outer membrane protein from Leptospira interrogans characterized in the C-terminus domain which play an important role in the infection and immunological responses of leptospirosis. [1,2]. Phylogenetic tree was constructed for Loa22 by comparing it with Lipoproteins (LipL 71, 45,
41, 31, 32, 21 and 48) and with outer membrane proteins (OmpL 1, 47, 54, 36, 37). The comprising lineages of Loa 22 have largely varying rates of evolution. In the present study the physicochemical properties of Loa22 were assessed. The structure of Loa22 was predicted by threading method in RaptorX server and the structure was energy minimized and validated by using SAVES server. The stereochemical quality and the protein backbone conformations were done by Ramachandran Plot analysis. Four sequential and conformational B cell epitopes of Loa22 were found in Ellipro server and mapped to find the sequential epitopes. The same B cell linear epitopes was also predicted by ABC Pred, BepiPred 1.0 server. All these epitopes were found to be in higher values. The results of the present study will help to elucidate the function of Loa22 and epitope-based vaccine development for Leptospirosis.
Key words: Leptospira, Lipoprotein, OmpA, outer membrane protein, Loa22, Phylogenetic analysis, B-cell epitope,
List of abbreviations:
SAVES: Structural Analysis and Verification Server; MEGA: Molecular Evolutionary Genetic Analysis; PI: Protrusion Index; LBP: Local Bootstrap values; ACC: Auto Cross Covariance NEFF: Number of Effective sequence homologs.
—————————— ——————————
Loa22 an OmpA like Outer membrane protein from Leptospira interrogans characterized by C-terminus domain invokes leptospiral pathogenesis. In other bacteria, OmpA acts as a multifunctional protein involved in cell adhesion, tissue invasion [3, 4], immune evasion [5], and induction of the immune response [6]. Leptospirosis is one of the most widespread zoonotic diseases in the world which is caused by the pathogenic bacteria Leptospira [7]. It is also known as Weil’s syndrome, and the clinical manifestations of leptospirosis occur when humans acquire the pathogen Leptospira from animals via skin or gastrointestinal contact with water, food or soil. Clinical symptoms of leptospirosis include high fever, bleeding, and renal failure. The major target of Leptospira in the kidney is the renal proximal tubular cells, and the pre- treatment with Leptospira outer membrane proteins, (OMPs) which leads to tubule interstitial nephritis and acute renal malfunction. [8] Mortality ranges from 10-15% with Weil’s disease to 70% in the case of pulmonary hemorrhage![]()
1 School of Biological Sciences, Madurai Kamaraj University, Madurai-
625021, Tamil Nadu, India.
2 Senior Professor & Head, Department of Psychopharmacology, National Institute of Mental Health and Neuro Sciences (NIMHANS), Hosur Road, Bangalore- 560029, Karnataka, India.
* Corresponding author
Telephone Number: +91-080- 26995108.
vaccine candidates have been problematic [19, 25]. Loa22 is
highly conserved among pathogenic Leptospira species; Loa22
syndrome (SPHS) [9-11]. Leptospirosis is endemic in most of the southern states of India like Kerala, Tamil Nadu and certain parts of Andhra Pradesh [12-14].
Leptospira genus is sub-classified into 18 genomospecies that included both saprophytic and pathogenic species [7, 15]. Based on serological methods, approximately 300 serovars have been identified; of which more than 200 are pathogenic [7, 16, 17]. The availability of genome sequence data for different Leptospira strains drives the discovery of new diagnostic tools and vaccines for Leptospirosis [18]. The major problem associated with Leptospirosis is the diagnosis of the disease, as it shows multiple symptoms; very often it is been confused with other diseases.
A number of leptospiral outer membrane proteins (OMPs) and lipoproteins have been characterized including OmpL1 [19], LipL41 [20], LipL36 [21], LipL32 [22], LipL21 [23], LipL46 [24], LenA [25], Loa22 [23] and Omp52 [19]. However their performance in diagnostic assays for acute leptospirosis or as is essential for virulence of L. interrogans in animal model and represents the first genetically defined virulence factor in
Leptospira species. More recent studies have revealed that
Loa22 may play an important role in the infection and immunological responses of Leptospirosis. Some outer membrane proteins of Leptospiral lipoproteins LipL32 (also
IJSER © 2015 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 6, Issue 3, March-2015 478
ISSN 2229-5518
known as Hap1), LipL41 and the porin OmpL1, have been
found to be protective immunogens that are conserved among various pathogenic leptospires. [26, 27].
Considering the potential importance of the outer membrane protein Loa22 as an epitope-based vaccine candidate, we examined the evolutionary relationship of Loa22 with several outer membrane and Lipoproteins, which showed comprising lineages. Given the potential of the Loa22 proteins as diagnostic antigens and vaccine candidates, the evolutionary relationship of Loa22 was examined, followed by protein threading and mapping of B-cell epitopes from the conserved region of the alignment to develop a vaccine candidate for Leptospirosis.
A total of 7 sequences of Lipoproteins such as LipL 71, LipL
45, LipL 41, LipL 31, LipL 32, LipL 21 and LipL 48 (Table. 1)
and 5 outer membrane proteins such as Omp L 1, Omp L 47, Omp L 54, Omp L 36, Omp L 37 (Table. 2)
from the species of L. interrogans serogroup
Icterohaemorrhagiae serovar copenhageni (strain Fiocruz L1-
130) and Loa22 from L. interrogans serovar Grippotyphosa (Table. 3) were retrieved from protein knowledgebase (UniProt KB) (http://www.uniprot.org/uniprot/). The retrieved sequences were subjected to Multiple Sequence Alignment with ClustalW algorithm, implemented in Molecular Evolutionary Genetic Analysis (MEGA 5.2.2) (http://www.megasoftware.net) by using distance matrix and was trimmed to consensus. Maximum Likelihood (ML) trees were constructed with 1000 bootstraps at uniform divergence rates with the distance ‘p’ as the evolutionary model and with a data subset to use with missing data treatment [28]. Posterior probability and the conserved regions among the closely related sequences were aligned with MEGA 5.2.2. (Figure 1). Maximum likelihood method selects the most suitable substitution model. The purpose of the molecular phylogenetic tree was to estimate the relationships among the species represented by the sequences and to understand the relationships among the sequences themselves without regard to the host species.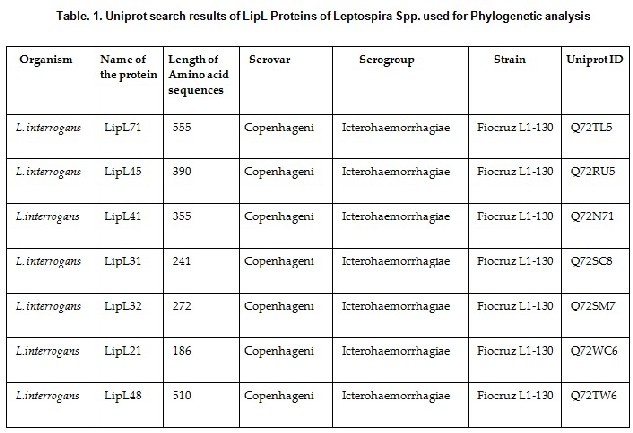
IJSER © 2015 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 6, Issue 3, March-2015 479
ISSN 2229-5518
Figure 1: Diagram showing the phylogenetic comparision of the Putative Lipoprotein Loa 22 which has a predicted OmpA domain based on sequence identity
Physicochemical analysis including molecular weight, theoretical pI, amino acid composition, instability index, aliphatic index, and grand average of hydropathicity (GRAVY) of Loa22 was analysed by using ProtParam tool (http://web.expasy.org/ protparam/). [34]. Prediction of subcellular localisation of the protein was carried out by CELLOv.2.5 [35].The secondary structure of the protein was studied by using PSIPRED server [36] BLAST [38] from NCBI was used to compare the query sequence with the database sequence and its homologues. Conserved domains [38] were also analysed from CDD of BLAST. Signal peptide regions were also predicted by using
Phobious signal predictor [39]. SOSUI [40] was used
to characterize whether the protein is soluble or transmembrane in nature. Interproscan [41] analysis of the query protein for domain study showed that it has a match of outer membrane protein, OmpA / MotB, C- terminal (IPR006665) of Omp A like superfamily. A collection of protein fingerprints by PRINTS [42] was used to study the tentative functional assignments for the selected query protein.
Since there is no any structure in the form of X- Ray crystallographic data from PDB all these above works were done only with the primary sequence information which was available from Uniprot for Loa22.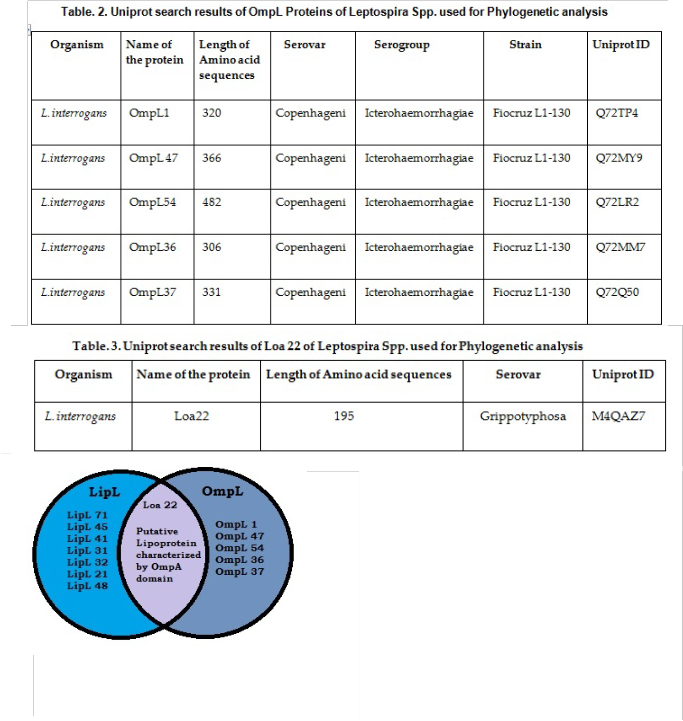
The retrieved Loa 22 sequence (Uniprot Id: M4QAZ7) was submitted into the RaptorX server [29] (http://raptorx.uchicago.edu/) to derive the 3-D structure. The modelled protein structure was viewed by using Swiss-PdbViewer (http://www.expasy. org/spdbv/) and the individual residues were collected using 100 cycles of steepest descent algorithm carried out in GROMOS96 [30] until the side chain interactions in the vicinity is readjusted and brings up lower potential energy and becomes more stable.
IJSER © 2015 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 6, Issue 3, March-2015 480

ISSN 2229-5518
The minimized energy models were assessed by
PROCHECK [31] for analysing the stereo chemical quality and residue by residue geometry of the modelled structure by submitting the co-ordinate file to Structural Analysis and Verification Server (SAVES) http://nihserver.mbi.ucla. edu/SAVES/). The value of the predicted Loa22 model was visualized and examined using PYMOL [32, 33].
Linear B-cell epitopes were mapped from the generated three dimensional structure of Loa22 and was chosen with two different algorithms: ABCPred and BepiPred. ABCPred uses a recurrent neural network to predict B-cell epitopes (http://www.imtech.res.in/raghava/abcpred/) [43,
44]. The amino acid length of 16 and the scoring threshold of 0.8 were set to predict B-cell epitopes in ABCPred. The epitope prediction in BepiPred is based on hidden Markov model (http://www.cbs.dtu.dk/services/BepiPred/) and and propensity scale method [45, 46]. The value, 0.35 was set as the threshold value, because at this value, the sensitivity/ specificity of the predictions will be maximized in BepiPred. BepiPred analyzes each amino acid independently and does not have a minimum or maximum number of amino acids to predict an epitope.
B-cell epitopes were predicted using Ellipro which is an Antibody Epitope Prediction server (http://tools.Immune epitope.org/tool s/ElliPro/iedbinput) [47, 48]. Ellipro, with the best algorithm to predict linear epitopes from 3-D structures when compared to six other software programs that predict linear epitopes [47]. The default threshold value was set at 0.8. The predicted epitopes was additionally verified in VaxiJen to predict the probability of an antigen (http://www.ddgpharmfac
.net/vaxijen/ VaxiJen/VaxiJen.html/) [49], with a threshold
of 0.4. VaxiJen uses an alignment free approach for antigen prediction and works on an auto cross covariance (ACC) transformation of protein sequences into uniform vectors of principal amino acid properties.
The phylogenetic tree was constructed using 7 Lipoprotein sequences (Table. 1), and 5 outer membrane protein sequences (Table. 2), all the sequences were retrieved from the species of L. interrogans serogroup Icterohaemorrhagiae serovar copenhageni (strain Fiocruz L1-130), and Loa22 from L. interrogans serovar Grippotyphosa (Table. 3) ) from the Uniprot Knowledge database. NCBI and tBLASTn were used to search the homologous sequences. 7 Lipoproteins and 5 Omps were compared with Loa 22 and its homologous sequences; phylogenetic analysis was performed and the evolutionary tree inferred by the ML method is shown in Figure 2 and Figure 3. Phylogenetic analyses result showed that, there are proteins including
Loa22 which clustered into the same
group with Lipoproteins and outer membrane proteins,
belonging to the species of L. interrogans serogroup Icterohaemorrhagiae serovar copenhageni (strain Fiocruz L1-130). The percentage of replicate trees in which the associated taxa clustered together in the 1000 bootstrap replicates test. Evolutionary analyses were conducted in MEGA5. The analysis involved 6 and 8 amino acid sequences separately. All positions containing gaps and missing data were eliminated. There were a total of 195 and 161 positions in the final dataset.
The ProtParam results exhibited the physicochemical parameters of the protein of Loa 22. It was predicted to have 195 Aminoacids (Table 4) with the molecular weight of 20884.5 Daltons and the theoretical Isoelectric point (PI) of 8.56. The instability index (II) was computed to be
44.64, implies that the sequence of Loa 22 is stable. The
maximum number of residues in the sequence were found to be alanine (13.3%) followed by serine (8.7 %) and whereas glycine, glycine and leucine equally shares 7.2% (Table.4). The sequence has about 24 negatively charged residues (Aspartic acid + Glutamic acid) and 26 positively charged residues (Arginine + Lysine). The amino-acid composition revealed that the protein has 2951 atoms comprising Carbon (922), Hydrogen (1479), Nitrogen (257), Oxygen (291) and sulfur (2). Thus C H N O S -922
1479 257 291 2 has been arrived as the molecular formula
for the protein Loa 22. The aliphatic index was calculated as 78.21. The grand average of hydropathicity (GRAVY) was calculated to be -0.430. This low range of value indicates that the protein has better interaction with water. The subcellular localization of protein using CELLO predicted that the query protein is a periplasmic protein. SOSUI results predicted that it is a membrane protein with hydrophobicity of -0.429744. The secondary structural analysis of the protein by PSIPRED showed that the random coil was found to be more frequent (48.72%) followed by extended strand to be the least frequent (21.03) helix and was found α to be as
30.26%.(Figure 4). The CDD BLAST results showed the
query protein comes under OmpA_C- like super family (Figure 5) and failed to give any more information regarding the nature of domain. The Interproscan analysis showed that the query protein might be that it has a match of Outer membrane protein, OmpA / MotB, C- terminal (IPR006665) of Omp A like superfamily (Table 5; Figure 5). Four motifs were recognized by the PRINTS server (Table 6). Phobious signal predictor also showed that the protein with signal peptide from 1 to 23 sequences which includes N region (1-7), H regions (8-18), C regions (19-23) and non cytoplasmic regions (23- 195). (Data not shown)
IJSER ©
International Journal of Scientific & Engineering Research, Volume 6, Issue 3, March-2015 481
ISSN 2229-5518
Figure. 4: Secondary structural elements of Loa 22, showing the alpha helix, beta strand and the coils of the amino acid target sequence.

Figure.2: The evolutionary history was inferred by using the Maximum Likelihood method based on the JTT matrix-based model [28]. The bootstrap consensus tree was inferred from 1000 replicates. The Branches corresponding to partitions reproduced in less than 50% bootstrap replicates are collapsed. The percentage of the replicate trees in which the clustered taxa are shown in branches. This analysis involves 6 amino acid sequences. Evolutionary analyses were conducted in MEGA5 [28]. The bootstrap value 43 shows the uniform support and indicating that the clade is a group. The scale bar represents branch length (number of amino acid substitutions/100 residues.

Figure.3: Phylogenetic tree with 7 sequences of LipL proteins were compared with Loa22. The reflection nodes with BP value of 71 indicating that
Loa22 is belonging to the group.
IJSER © 2015 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 6, Issue 3, March-2015 482
ISSN 2229-5518
Sequence Domain Number 1 | M4QAZ7_LEPIR Region 1 - 195 | |
Classification Level | Classification | E Value |
Superfamily | Omp A like | SSF103088 |
Family | OmpA/Mot B, C- terminal | IPR006665 |
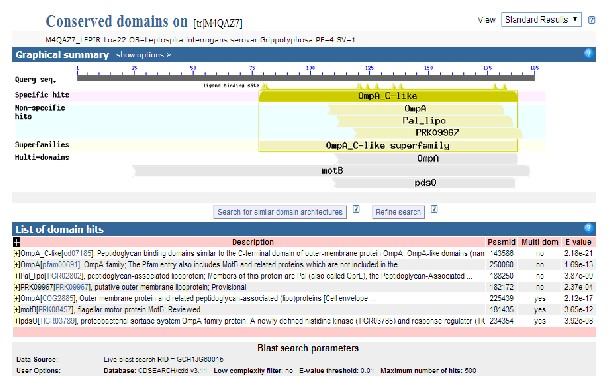
Figure.5: CDD results of Loa 22 showing the protein comes under OmpA_C- like super family
Given an input sequence, RaptorX predicted its tertiary
Normally single template is being used to generate protein structures by homology modelling and protein threading. But if the sequence is distantly related to the template or if the template does not cover the complete sequence, protein modelling will not be useful. The threading based-prediction is based on the predicted structure properties, such as the predicted secondary structures or the predicted residue burial status [50]. Threading based prediction was done in RaptorX server which uses a non-linear scoring function to combine homologous information with structural information for the given template-sequence alignment [50]. The protein was modelled by protein threading.
structure as well as solvent accessibility and disordered regions (Figure.6). RaptorX assigned the confidence score which is based on P-value and uGDT (unnormalized Global Distance Test). P-value measures the relative quality of the model. For alpha proteins, P-value less than
10^-3 is a good indicator. For beta proteins, P-value less
than 10^-4 is a good indicator. uGDT measures the absolute model quality. uGDT greater than 50 is a good indicator.
The NEFF (Number of Effective sequence
homologs) score which was ranging from 1 to 20 for the predicted structure was estimated by PROCHECK. The results obtained from PROCHECK [51] were evaluated for protein backbone conformations by Ramachandran Plot
[52, 53].
IJSER © 2015 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 6, Issue 3, March-2015 483
ISSN 2229-5518
The phi-psi torsion angle for 87.5 % of
residues of Loa22 are in the most favourable region (A, B and L); 9.5%, 2.4% and 0.6 % in additionally allowed, generously allowed and disallowed regions respectively (a, b, l, p) indicating that Loa22 model is stereo chemically good and the model derived from RaptorX was of higher quality in terms of protein folding. (Figure. 7)
Surface accessibility and fragment flexibility are important features for predicting antigenic epitopes. In addition, the existence of regions with high hydrophobicity also provides strong evidence for epitope identification. Based on the sequence properties by physicochemical analysis, two different epitope prediction software programs (ABCPred and BepiPred) were utilized to predict the most immunogenic linear B-cell epitopes on the surface of the Leptospiral OmpA like protein Loa22.
ABCPred and BepiPred predicted 8 different overlapping and potentially immunogenic regions within Loa 22 respectively (data not shown). ABCPred is able to predict epitopes with approximately 66% accuracy using the recurrent neural network [43]. ABCPred assigns scores between 1 and 0 for each epitope it predicts. An epitope shows score closer to 1 can be taken and score closer to 0 is not suitable for an epitope.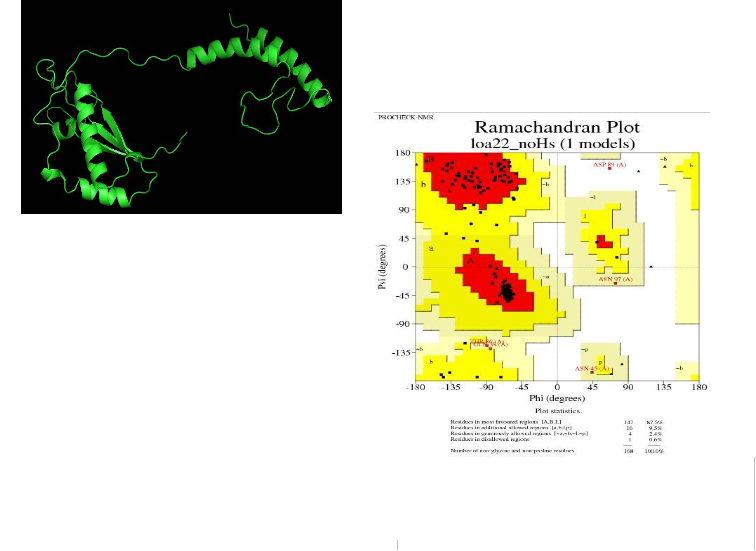
Four B- cell epitopes of Loa 22 were mapped from the predicted 3-D structure of Loa22 by using Ellipro (Figure. 8). These epitopes covers (EP1: 1-6), (EP2: 19-28), (EP3: 125-131), (EP4: 172-177) (Table 7). Ellipro predicts epitope with Protrusion Index (PI) value which is percentage of the protein atoms enclosed in the ellipsoid, at which the residue first becomes lying outside the ellipsoid; whereas all the residues which were lying 90% outside the ellipsoid had the PI value 9, i.e., 0.9 in Ellipro. This gives information of amino acids lying outside the ellipsoid. The prediction of peptides is vital not only for diagnostics but also for vaccines. It became clear that the small segments of protein called the antigenic determinants or the epitopes are sufficient for eliciting the desired immune response. Based on the threshold value, all the predicted epitopes are antigenic nature. All these results can be used to benefit immunotherapy and also for the development of
diagnostic based approaches for Leptospirosis.
Figure. 6: Three dimensional structure of Loa22 of L. interrogans serovar
Grippotyphosa (M4QAZ7) which was energy minimized and visualized by
Pymol shown in cartoon representation
Figure. 7: The results obtained from PROCHECK were evaluated for protein backbone conformations by Ramachandran Plot. The phi-psi torsion angle for 87.5 % of residues of Loa22 are in the most favour- able region (A, B and L); 9.5%, 2.4% and 0.6 % in additionally allo-
wed, generously allowed and disallowed regions respectively (a, b, l, p)
IJSER © 2015 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 6, Issue 3, March-2015 484
ISSN 2229-5518
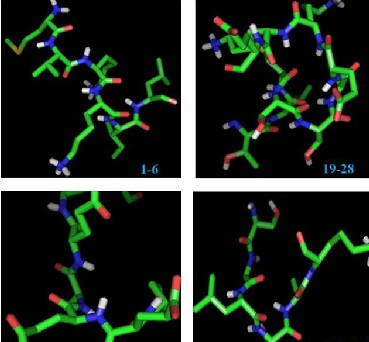
Figure. 8: B cell epitopes modelled and visualised using PYMOL shown in stick from the modelled three dimensional structure of Loa22
In this study, we have a better understanding of the 2D and 3D structure of Loa22. Ramachandran plot analysis confirmed stereochemical model and have predicted four B cell epitopes (EP1: 1-6), (EP2: 19-28), (EP3: 125-131), (EP4: 172-177). The
goodness factor (𝐺-factor) based on the observed distribution of
stereochemical parameters (main chain bond angles, bond
length, and phi-psi torsion angles) returned accurate values for
a reliable model indicating that Loa22 model is stereo
chemically good. The most innovative areas in the vaccine as on today are the DNA vaccines for Leptospira. Until now, four OMPs (OmpL1, LipL32, LipL21 and LigA) were evaluated for their DNA vaccine efficacy [54]. Thus all these datas generated for Loa22 can be used to benefit immunotherapies but still further studies are required based on these bioinformatic approaches.
Acknowledgement
The authors thank Department of Pschycopharmacology, NIMHANS, Bangalore and School of Biological Sciences, Madurai Kamaraj University, Madurai-625021, Tamil Nadu, India for the necessary work and software facilities.
References
[1] Koizumi N and Watanabe H. (2003). Molecular cloning and characterization of a novel leptospiral lipoprotein with OmpA domain. FEMS Microbiol Lett, 226: 215–219.
[2] Gamberini M, Gomez RM, Atzingen MV, Martins EA, Vasconcellos SA, Romero EC and Leite LC, et al. (2005). Whole-genome analysis of Leptospira interrogans to identify potential vaccine candidates against leptospirosis. FEMS Microbiol Lett, 244: 305–313.
[3] Fagan RP and Smith SG. (2007). The Hek outer membrane protein of Escherichia coli is an auto-aggregating adhesin and invasion. FEMS Microbiol Lett, 269: 248–255.
[4] Teng CH, Xie Y, Shin S, Di Cello F, Paul-Satyaseela M, Cai M and Kim KS. (2006). Effects of OmpA deletion on expression of type 1 fimbriae in Escherichia coli K1 strain RS218 and on the association of E. coli with human brain microvascular endothelial cells. Infect Immun 74: 5609–5616.
[5] Wooster DG, Maruvada R, Blom AM and Prasadarao NV. (2006).
Logarithmic phase Escherichia coli K1 efficiently avoids serum killingby promoting C4bp-mediated C3b andC4b degradation. Immunol. 117: 482–493.
[6] Torres AG, Li Y, Tutt CB, Xin L, Eaves-Pyles T and Soong L. (2006).
Outer membrane protein A of Escherichia coli O157:H7 stimulates dendritic cell activation. Infect Immun, 74: 2676–2685.
[7] Levett P. N., Branch S. L., Whittington C. U., Edwards C. N., Paxton H. (2001). Two Methods for Rapid Serological Diagnosis of Acute Leptospirosis. Clin. Diagn. Lab. Immunol. 8:349–351
[8] Dolhnikoff, M., Mauad, T., Bethlem, E. P. & Carvalho, C. R. (2007).
Pathology and Pathophysiology of Pulmonary Manifestations in
Leptospirosis. Braz J Infect Dis. 11: 142–148.
[9] Gouveia EL, Metcalfe J, De Carvalho AL, Aires TS, Villasboas- Bisneto JC, Queirroz A, Santos AC, Salgado K, Reis MG, Ko AI (2008). Leptospirosis-associated Severe Pulmonary Hemorrhagic Syndrome, Salvador. Brazil Emerg Infect Dis. 14:505–508.
[10] McBride AJ, Athanazio DA, Reis MG, Ko AI (2005).
Leptospirosis. Curr Opin Infect. Dis 18:376–386.
[11] Segura ER, Ganoza CA, Campos K, Ricaldi JN, Torres S, Silva H, Cespedes MJ, Matthias MA, Swancutt MA, Lopez Linan R, et al. (2005). Clinical spectrum of pulmonary involvement in leptospirosis in a region of endemicity, with quantification of leptospiral burden. Clin Infect Dis. 40(3):343–351.
[12] Koteeswaran AS (2006). Seroprevalence of leptospirosis in man and animals analysis of the gene encoding LipL32 of Leptospira in Tamil Nadu. Indian J Med Microbiol, 24: 329-331.
[13] Balakrishnan G, Govindarajan R, Meenambigai TV, Jayakumar V, Manohar MB (2008). Diagnosis of leptospirosis by recombinant antigen: Seroprevalence of animal based single serum dilution ELISA. Indian Vet J. 85: 227- 228
[14] Abhinay G, Joseph S, Ambily R (2012). Sero prevalence of canine leptospirosis. Indian Vet J 89: 72-73.
[15] Lin MH, Chang Y.C, Hsiao CD, Huang SH, Wang MS, (2013).
LipL41, a Hemin Binding Protein from Leptospira santarosai serovar
Shermani. PLoS ONE 8:12.
[16] Faine S, Adler B, Bolin C, Perolat P (1999) Leptospira and
leptospirosis, 2nd ed. MediSci. Melbourne, Australia.
[17] Bharti A, Nally J, Ricaldi J, Matthias M, Diaz M, Lovett M, Levett P, Gilman R, Willig M, Gotuzzo E, Vinetz J (2003) Leptospirosis: A Zoonotic Disease of Global Importance. Lancet Infect Disease 3: 757–
771.
[18] Adler B, De La Pena Moctezuma A (2010) Leptospira and
Leptospirosis. Vet Microbiol 140: 287–296.
[19] Haake DA, Mazel MK, Mccoy AM, Milward F, Chao G, Matsunaga J, Wagar EA (1999) Leptospiral Outer Membrane Proteins Ompl1 and Lipl41 Exhibit Synergistic Immunoprotection. Infect Immun 67:6572-
6582.
[20] Shang ES, Summers TA, Haake DA (1996). Molecular cloning and sequence analysis of the gene encoding LipL41, a surface-exposed lipoprotein of pathogenic Leptospira species. Infect Immun 64:2322–
2330.
[21] Haake DA, Chao G, Zuerner R, Barnett JK, Barnett D, Mazel M,
Matsunaga J, Levett PN, Bolin CA (2000). The leptospiral major outer membrane protein LipL32 is a lipoprotein expressed during mammalian infection. Infect Immun 68: 2276–2285.
[22] Cullen PA, Xu XY, Matsunaga J, Sanchez Y, Ko AI, Haake DA, Adler B (2005). Surface ome of Leptospira spp. Infect Immun73:4853–
4863.
[23] Matsunaga J, Barocchi MA, Croda J, Young TA, Sanchez Y, Siqueira I, Bolin CA, Reis MG, Riley LW, Haake DA, Ko AI (2003) Pathogenic Leptospira species express surface-exposed proteins belonging to the bacterial immunoglobulin superfamily. Mol Microbiol 49: 929–945.
IJSER © 2015 http://www.ijser.org
International Journal of Scientific & Engineering Research, Volume 6, Issue 3, March-2015 485
ISSN 2229-5518
[24] Verma A, Brissette CA, Bowman AA, Shah ST, Zipfel PF, Stevenson B (2010) Leptospiral Endostatin-Like Protein A is A Bacterial Cell Surface Receptor for Human Plasminogen. Infect Immun 78:2053–
2059.
[25] Guerreiro H, Croda J, Flannery B, Mazel M, Matsunaga J, Reis MG, Levett PN, Ko AI, Haake DA (2001) Leptospiral proteins recognized during the humoral immune response to leptospirosis in humans. Infect Immun 69:4958–4968.
[26] Shang, E.S., Summers, T.A. and Haake, D.A. (1996) Molecular cloning and sequence analysis of the gene encoding LipL41, a surface exposed lipoprotein of pathogenic Leptospira species. Infect. Immun. 64, 2322^2330.
[27] Branger, C., Sonrier, C., Chatrenet, B., Klonjkowski, B., Ruvoen- Clouet, N., Aubert, A., Andre¤-Fontaine, G. and Eloit, M. (2001) Identi¢cation of the hemolysis-associated protein 1 as a cross- protective immunogen of Leptospira interrogans by adenovirus- mediated vaccination. Infect. Immun. 69, 6831^6838.
[28] Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance and Maximum Parsimony Methods. Mol Biol Evol 28: 2731-2739.
[29] Morten KM, Haipeng WH, Sheng WS, Jian J, Zhiyong WZ, Hui LH, Jinbo XJ (2012) Template-Based Protein Structure Modelling Using the RaptorX Web Server. Nature 7:1511–1522.
[30] Bonvin AMJJ, Mark AE, Van Gunsteren WF (2000) The GROMOS96
benchmarks for molecular simulation. Comput phys commun 128:
550–557.
[31] Unit M, Street G (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr
26:283–291.
[32] Schrodinger LLC (2010). The PyMOL Molecular Graphics System, Version~1.3r1.
[33] DeLano WL (2002) The PyMOL Molecular Graphics System, http://www.pymol.org
[34] C. J. A. Sigrist, E. de Castro, L. Cerutti et al., (2013). “New and continuing developments at PROSITE,” Nucleic Acids Research, vol.
41, no. 1, pp. D344–D347.
[35] Yu, C.S., Lin, C.J., Hwang, J.K., (2006). Predicting subcellular localization of protein for gram negative bacteria by support vector machine based on polypeptide composition. Protein Science 13:1402-
1406.
[36] Jones DT. (1999). Protein secondary structure prediction based on position specific scoring matrices. J. Mol. Biol. 292: 195-202.
[37] Altschul, S.F., Gish, W., Miller, W., Myers, E.W. & Lipman, D.J.
(1990) "Basic local alignment search tool." J. Mol. Biol. 215:403-410. [38] Marchler-Bauer A et al. (2011), "CDD: a Conserved Domain Database
for the functional annotation of proteins", Nucleic Acids Res.39 (D) 225-
9.
[39] Lukas Käll, Anders Krogh and Erik L. L. Sonn hammer (2004) A Combined Transmembrane Topology and Signal Peptide Prediction Method. Journal of Molecular Biology, 338(5):1027-1036.
[40] Hirowaka, Y., Boon-Chieng-S. ,Mitaku, S., (1998). Classification and secondary structure prediction system for membrane proteins. Bioinformatics., 14:378-379.
[41] Zdobnov, E.M., Rolf, A., (2001). Interproscan –an integration platforms for the signature recognition methods in Interpro. Bioinformatics 17:847-848.
[42] Attwood, T. K., (2002).The PRINTS Database a resource for identification of protein families. Brief. Bioinform.3: 252-263.
[43] Pirovano W, Heringa J (2010) “Protein secondary structure prediction. Method Mol Biol 609: 327–348.
[44] Saha S, Raghava GPS (2007) Prediction of neurotoxins based on their function and source. In Silico Biol 7: 0025
[45] Larsen JEP, Lund O, Nielsen M (2006) “Improved method for predicting linear B-cell epitopes.” Immunome Res 2: 2.
[46] Kringelum JV et al. (2012) Reliable B cell epitopes predictions: impacts and method development and improved benchmarking. PLoS Comput Biol 8:1002829.
[47] Ponomarenko JV, Bui HH, Li W, Fussender N, Bourne PE, Sette A, Peters B (2008) Ellipro: A New Structure-Based Tool for the prediction of Antibody Epitopes. BMC Bioinformatics. 2: 9:514.
[48] Ponomarenko JV, Regenmortel Marc HV (2009) B-cell epitope prediction. Struct Bioinfo 35: 849-881.
[49] Doytchinova IA, Flower DR (2007). VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics 8:4.
[50] Morten KM, Haipeng WH, Sheng WS, Jian J, Zhiyong WZ, Hui LH, Jinbo XJ (2012) Template-Based Protein Structure Modelling Using the RaptorX Web Server. Nature 7:1511–1522.
[51] Unit M, Street G (1993) PROCHECK: a program to check the
stereochemical quality of protein structures. J Appl Crystallogr
26:283–291.
[52] Ramachandran GN, Ramakrishnan C, Sasisekharan V (1963) "Stereochemistry of Polypeptide Chain Configurations". J Mol Biol 7:
95–9.
[53] Ramachandran GN, Sasishekaran V (1968) Conformation of polypeptides and proteins. Adv Prot Chem. 23: 283-437.
[54] Crit Rev Microbiol, Early Online: 1–17, (2013) Informa Healthcare
USA, Inc.
IJSER © 2015 http://www.ijser.org